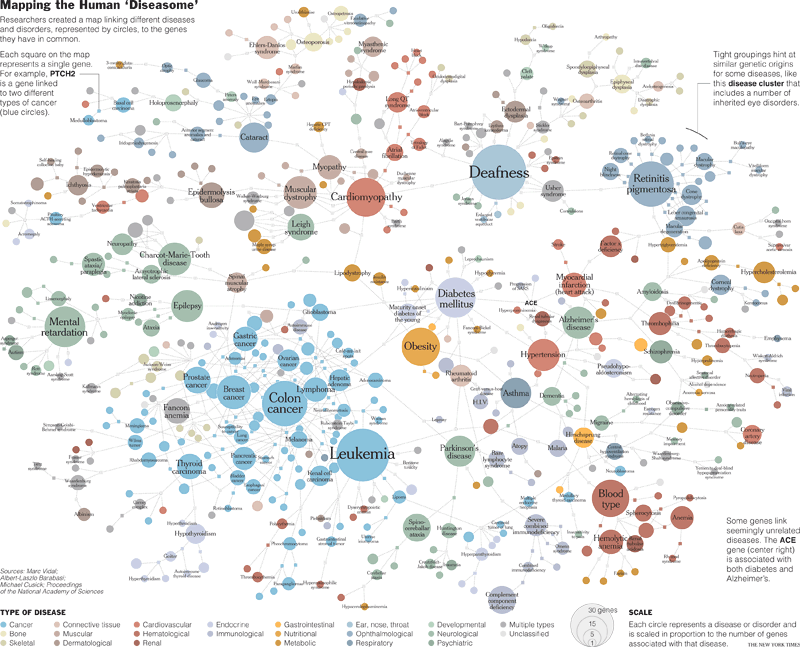
Results
Construction of the Diseasome.
We constructed a bipartite graph consisting of two disjoint sets of nodes. One set corresponds to all known genetic disorders, whereas the other set corresponds to all known disease genes in the human genome (
Fig. 1). A disorder and a gene are then connected by a link if mutations in that gene are implicated in that disorder. The list of disorders, disease genes, and associations between them was obtained from the Online Mendelian Inheritance in Man (OMIM; ref.
18), a compendium of human disease genes and phenotypes. As of December 2005, this list contained 1,284 disorders and 1,777 disease genes. OMIM initially focused on monogenic disorders but in recent years has expanded to include complex traits and the associated genetic mutations that confer susceptibility to these common disorders (
18). Although this history introduces some biases, and the disease gene record is far from complete, OMIM represents the most complete and up-to-date repository of all known disease genes and the disorders they confer. We manually classified each disorder into one of 22 disorder classes based on the physiological system affected [see
supporting information (SI) Text, SI Fig. 5, and SI Table 1 for details].

Fig. 1.
Construction of the diseasome bipartite network. (
Center) A small subset of OMIM-based disorder–disease gene associations (
18), where circles and rectangles correspond to disorders and disease genes, respectively. A link is placed between a disorder and a disease gene if mutations in that gene lead to the specific disorder. The size of a circle is proportional to the number of genes participating in the corresponding disorder, and the color corresponds to the disorder class to which the disease belongs. (
Left) The HDN projection of the diseasome bipartite graph, in which two disorders are connected if there is a gene that is implicated in both. The width of a link is proportional to the number of genes that are implicated in both diseases. For example, three genes are implicated in both breast cancer and prostate cancer, resulting in a link of weight three between them. (
Right) The DGN projection where two genes are connected if they are involved in the same disorder. The width of a link is proportional to the number of diseases with which the two genes are commonly associated. A full diseasome bipartite map is provided as
SI Fig. 13.
Starting from the diseasome bipartite graph we generated two biologically relevant network projections (
Fig. 1). In the “human disease network” (HDN) nodes represent disorders, and two disorders are connected to each other if they share at least one gene in which mutations are associated with both disorders (
Figs. 1 and
2 a). In the “disease gene network” (DGN) nodes represent disease genes, and two genes are connected if they are associated with the same disorder (
Figs. 1 and
2 b). Next, we discuss the potential of these networks to help us understand and represent in a single framework all known disease gene and phenotype associations.

Fig. 2.
The HDN and the DGN. (
a) In the HDN, each node corresponds to a distinct disorder, colored based on the disorder class to which it belongs, the name of the 22 disorder classes being shown on the right. A link between disorders in the same disorder class is colored with the corresponding dimmer color and links connecting different disorder classes are gray. The size of each node is proportional to the number of genes participating in the corresponding disorder (see key), and the link thickness is proportional to the number of genes shared by the disorders it connects. We indicate the name of disorders with >10 associated genes, as well as those mentioned in the text. For a complete set of names, see
SI Fig. 13. (
b) In the DGN, each node is a gene, with two genes being connected if they are implicated in the same disorder. The size of each node is proportional to the number of disorders in which the gene is implicated (see key). Nodes are light gray if the corresponding genes are associated with more than one disorder class. Genes associated with more than five disorders, and those mentioned in the text, are indicated with the gene symbol. Only nodes with at least one link are shown.
Properties of the HDN.
If each human disorder tends to have a distinct and unique genetic origin, then the HDN would be disconnected into many single nodes corresponding to specific disorders or grouped into small clusters of a few closely related disorders. In contrast, the obtained HDN displays many connections between both individual disorders and disorder classes (
Fig. 2 a). Of 1,284 disorders, 867 have at least one link to other disorders, and 516 disorders form a giant component, suggesting that the genetic origins of most diseases, to some extent, are shared with other diseases. The number of genes associated with a disorder,
s, has a broad distribution (see
SI Fig. 6a ), indicating that most disorders relate to a few disease genes, whereas a handful of phenotypes, such as deafness (
s = 41), leukemia (
s = 37), and colon cancer (
s = 34), relate to dozens of genes (
Fig. 2 a). The degree (
k) distribution of HDN (
SI Fig. 6b ) indicates that most disorders are linked to only a few other disorders, whereas a few phenotypes such as colon cancer (linked to
k = 50 other disorders) or breast cancer (
k = 30) represent hubs that are connected to a large number of distinct disorders. The prominence of cancer among the most connected disorders arises in part from the many clinically distinct cancer subtypes tightly connected with each other through common tumor repressor genes such as
TP53 and
PTEN.
Although the HDN layout was generated independently of any knowledge on disorder classes, the resulting network is naturally and visibly clustered according to major disorder classes. Yet, there are visible differences between different classes of disorders. Whereas the large cancer cluster is tightly interconnected due to the many genes associated with multiple types of cancer (
TP53,
KRAS,
ERBB2,
NF1, etc.) and includes several diseases with strong predisposition to cancer, such as Fanconi anemia and ataxia telangiectasia, metabolic disorders do not appear to form a single distinct cluster but are underrepresented in the giant component and overrepresented in the small connected components (
Fig. 2 a). To quantify this difference, we measured the locus heterogeneity of each disorder class and the fraction of disorders that are connected to each other in the HDN (see
SI Text ). We find that cancer and neurological disorders show high locus heterogeneity and also represent the most connected disease classes, in contrast with metabolic, skeletal, and multiple disorders that have low genetic heterogeneity and are the least connected (
SI Fig. 7).
Properties of the DGN.
In the DGN, two disease genes are connected if they are associated with the same disorder, providing a complementary, gene-centered view of the diseasome. Given that the links signify related phenotypic association between two genes, they represent a measure of their phenotypic relatedness, which could be used in future studies, in conjunction with protein–protein interactions (
6,
7,
19), transcription factor-promoter interactions (
20), and metabolic reactions (
8), to discover novel genetic interactions. In the DGN, 1,377 of 1,777 disease genes are connected to other disease genes, and 903 genes belong to a giant component (
Fig. 2 b). Whereas the number of genes involved in multiple diseases decreases rapidly (
SI Fig. 6d ; light gray nodes in
Fig. 2 b), several disease genes (e.g.,
TP53,
PAX6) are involved in as many as 10 disorders, representing major hubs in the network.
Functional Clustering of HDN and DGN.
To probe how the topology of the HDN and GDN deviates from random, we randomly shuffled the associations between disorders and genes, while keeping the number of links per each disorder and disease gene in the bipartite network unchanged. Interestingly, the average size of the giant component of 10
4 randomized disease networks is 643 ± 16, significantly larger than 516 (
P < 10
−4; for details of statistical analyses of the results reported hereafter, see
SI Text ), the actual size of the HDN (
SI Fig. 6c ). Similarly, the average size of the giant component from randomized gene networks is 1,087 ± 20 genes, significantly larger than 903 (
P < 10
−4), the actual size of the DGN (
SI Fig. 6e). These differences suggest important pathophysiological clustering of disorders and disease genes. Indeed, in the actual networks disorders (genes) are more likely linked to disorders (genes) of the same disorder class. For example, in the HDN there are 812 links between disorders of the same class, an 8-fold enrichment with respect to 107 ± 10 links obtained between the same set of nodes in the randomized networks. This local functional clustering accounts for the small size of the giant components observed in the actual networks.
Disease-Associated Genes Identify Distinct Functional Modules.
For several disorders known to arise from mutations in any one of a few distinct genes, the corresponding protein products have been shown to participate in the same cellular pathway, molecular complex, or functional module (
21,
22). For example, Fanconi anemia arises from mutations in a set of genes encoding proteins involved in DNA repair, many of them forming a single heteromeric complex (
23). Yet, the extent to which most disorders and disorder classes correspond to distinct functional modules in the cellular network has remained largely unclear. If genes linked by disorder associations encode proteins that interact in functionally distinguishable modules, then the proteins within such disease modules should more likely interact with one another than with other proteins. To test this hypothesis, we overlaid the DGN on a network of physical protein–protein interactions derived from high-quality systematic interactome mapping (
6,
7) and literature curation (
6). We found that 290 interactions overlap between the two networks, a 10-fold increase relative to random expectation (
P < 10
−6;
Fig. 3 a).
Fig. 3.
Characterizing the disease modules. (a) Number of observed physical interactions between the products of genes within the same disorder (red arrow) and the distribution of the expected number of interactions for the random control (blue) (P < 10−6). (b) Distribution of the tissue-homogeneity of a disorder (red). Random control (blue) with the same number of genes chosen randomly is shown for comparison. (c) The distribution of PCC ρ ijvalues of the expression profiles of each disease gene pair that belongs to the same disorder (red) and the control (blue), representing the PCC distribution between all gene pairs (P < 10−6). (d) Distribution of the average PCC between expression profiles of all genes associated with the same disorder (red) is also shifted toward higher values than the random control (blue) with the same number of genes chosen randomly (P < 10−6).
Genes associated with the same disorder share common cellular and functional characteristics, as annotated in the Gene Ontology (GO) (
24). If the HDN shows modular organization, then a group of genes associated with the same common disorder should share similar cellular and functional characteristics, as annotated in GO. To investigate the validity of this hypothesis, we measured the GO homogeneity of each disorder (see
SI Text ) separately for each branch of GO, biological process, molecular function, and cellular component, finding significant elevation of GO homogeneity with respect to random controls in all three branches (
SI Fig. 8).
Disease genes encoding proteins that interact within common functional modules should tend to be expressed in the same tissue. To measure this, we introduced the tissue-homogeneity coefficient of a disorder, defined as the maximum fraction of genes among those belonging to a common disorder that are expressed in a specific tissue in a microarray data set obtained for 10,594 genes across 36 healthy tissues (
25). We found that 68% of disorders exhibited almost perfect tissue-homogeneity (
Fig. 3 b), compared with 51% expected by chance (
P < 10
−5).
Finally, disease genes that participate in a common functional module should also show high expression profiling correlation (
26). The distribution of Pearson correlation coefficients (PCCs) for the coexpression profiles of pairs of genes associated with the same disorder was shifted toward higher values compared with that of a random control (
Fig. 3 c;
P < 10
−6, χ
2 test). Similarly, the average PCC over all pairs of genes within a given disorder shows a significant shift from the random reference (
Fig. 3 d), with a small but clearly distinguishable peak in the distribution around PCC ≈ 0.75. This peak corresponds to ≈33 disorders with average PCC > 0.6 for which all genes are highly coexpressed in most tissues, including Heinz body anemia (PCC = 0.935), Bethlem myopathy (PCC = 0.835), and spherocytosis (PCC = 0.656).
In summary, genes that contribute to a common disorder (
i) show an increased tendency for their products to interact with each other through protein–protein interactions, (
ii) have a tendency to be expressed together in specific tissues, (
iii) tend to display high coexpression levels, (
iv) exhibit synchronized expression as a group, and (
v) tend to share GO terms. Together, these findings support the hypothesis of a global functional relatedness for disease genes and their products and offer a network-based model for the diseasome. Cellular networks are modular, consisting of groups of highly interconnected proteins responsible for specific cellular functions (
21,
22). A disorder then represents the perturbation or breakdown of a specific functional module caused by variation in one or more of the components producing recognizable developmental and/or physiological abnormalities.
This model offers a network-based explanation for the emergence of complex or polygenic disorders: a phenotype often correlates with the inability of a particular functional module to carry out its basic functions. For extended modules, many different combinations of perturbed genes could incapacitate the module, as a result of which mutations in different genes will appear to lead to the same phenotype. This correlation between disease and functional modules can also inform our understanding of cellular networks by helping us to identify which genes are involved in the same cellular function or network module (
21,
22).
Centrality and Peripherality.
An early indication of the connection between the structure of a cellular network and its functional properties was the finding that in
Saccharomyces cerevisiae highly connected proteins or “hubs” are more likely encoded by essential genes (
15,
16). This prompted a number of recent studies (
27,
28) to formulate the hypothesis that human disease genes should also have a tendency to encode hubs. Yet, previous measurements found only a weak correlation between disease genes and hubs (
29), resulting in an important mystery: what is the role, if any, of the cellular network in human diseases? Are disease genes more likely to encode hubs in the cellular network?
Our initial analysis appears to support the hypothesis that disease genes, given their impact on the organism, display a tendency to encode hubs in the interactome (
27,
28), finding that disease related proteins have a 32% larger number of interactions (
6,
7) with other proteins (average degree) than the nondisease proteins (see
SI Fig 9) and that high-degree proteins are more likely to be encoded by genes associated with diseases than proteins with few interactions (
P = 1.6 × 10
−17;
Fig. 4 a). Next, we show, however, that despite this apparent correlation, the relationship between diseases and hubs hides deep differences between various disease genes.

Fig. 4.
Functional characteristics of disease and essential genes. (
a) The fraction of disease genes among those whose protein products that interact with
k other proteins. (
b) Venn diagram showing the relationship between the human genes studied in this work. (
c) The fraction of genes with lethal mouse phenotypes (essential genes) among those with mouse phenotypes that interact with
kother proteins. (
d) The same as in
a, but only for nonessential disease genes, i.e., excluding 398 proteins with lethal mouse phenotypes. (
e and
f) The fraction of essential genes (
e) and nonessential disease genes (
f) among those whose average PCC with other genes is 〈ρ〉. (
g and
h) The fraction of essential genes (
g) and nonessential disease genes (
h) among those whose transcript is expressed in
nT tissues. Gray horizontal lines in
a and
c–h indicate the global average. Error bars represent standard errors. Note that for some data points the error bars are smaller than the symbol size, and thus are not visible. In
a,
c, and
d gray symbols are the linearly binned data points, whereas color corresponds to the statistically more uniform log-binned data. For details of the significance analysis, see
SI Text .
When exploring whether disease genes encode hubs, we, and authors of other earlier studies (
27–
29), ignored the fact that some human genes are essential in early development and functional changes in these contribute to the high rate of first-trimester spontaneous abortions, which might be as much as 20% of recognized pregnancies. One strategy to explore the impact of this
in utero essential segment of human disease is to consider human orthologs of mouse genes that result in embryonic or postnatal lethality when disrupted by homologous recombination (Mouse Genome Informatics;
www.informatics.jax.org). All together, we find 1,267 such mouse lethal orthologs of human genes, of which 398 are associated with human diseases, representing 22% of all known human disease genes. This allows us to distinguish between two classes of human genes: 1,267 “essential genes” and 1,379 “nonessential disease genes,” the latter obtained by removing from the full list of 1,777 OMIM disease genes the 398 that are also essential (
Fig. 4b). Next, we show that these two classes of genes play quite different roles in the human interactome.
First, we find that essential proteins show a tendency to be associated with hubs (
P = 1.3 × 10
−17;
Fig. 4 c), displaying a much stronger trend than the one observed for all disease proteins (
Fig. 4 a). This raises an important question: Could the observed correlation between disease genes and hubs (
Fig. 4 a) be the sole consequence of the fact that a small fraction (22%) of disease genes is also essential? To address this question we measured the degree dependence of the nonessential disease proteins (
Fig. 4 d). Surprisingly, the correlation between hubs and disease proteins entirely disappears. Thus, the vast majority of disease genes (78%), those that are nonessential, do not show a tendency to encode hubs, indicating that the observed weak correlations between hubs and disease genes (
Fig. 4 a) was entirely due to the few essential genes within the disease gene class.
To carry on its basic functions, the cell needs to maintain the coordinated activity of important functional modules, driving in a relatively synchronized manner the expression patterns of the most important genes. Therefore, one expects that the expression pattern of both essential and disease genes will be synchronized with a significant number of other genes. To test this, we determined the average gene coexpression coefficient 〈ρ〉
i = Σ
j PCC
ij between an essential (or nonessential disease) gene
i and all other genes in the cell, calculating the PCC
ij values from healthy human tissue microarray measurements (
25). Confirming our expectation, for essential genes we find that genes that display high average coexpression 〈ρ〉 with all other genes are more likely to be essential than those that show small or negative 〈ρ〉 (
P = 1.7 × 10
−4;
Fig. 4 e). Surprisingly, however, nonessential disease genes show the opposite effect, being associated with genes whose expression pattern is anticorrelated or not-correlated with other genes, and underrepresented among the genes that are highly synchronized (〈ρ〉 > 0.2) (
P = 2.6 × 10
−8;
Fig. 4 f). Thus, the expression pattern of nonessential disease genes appears to be decoupled from the overall expression pattern of all other genes, whereas essential genes have a tendency to be coupled to the rest of the cell.
Finally, we asked whether housekeeping genes, expressed in all tissues, have a tendency to encode disease genes. We find that the more tissues in which a gene is expressed, the higher the likelihood that it will be essential (
P = 2.8 × 10
−16;
Fig. 4 g). The opposite is true for nonessential disease genes: they have a tendency to be expressed in a few tissues (
P = 1.4 × 10
−6;
Fig. 4 h). Similarly, we found that only 9.9% of housekeeping genes correspond to disease genes, compared with 13.5% of nonhousekeeping genes, a significant 36% difference (
P = 3.6 × 10
−6). In contrast, 59.8% of housekeeping genes annotated with mouse phenotype were essential, compared with 40.5% for nonhousekeeping genes (
P < 10
−4).
These results support the somewhat unexpected conclusion that nonessential disease genes are not associated with hubs (
27,
28), show smaller correlation in their expression pattern with the rest of the genes in the cell than expected from random, and have a tendency to be expressed in only a few tissues. Therefore, contrary to earlier hypotheses and our expectations, the vast majority of nonessential disease genes occupy functionally peripheral and topologically neutral positions in the cellular network. In stark contrast, essential genes are likely to encode hubs, show highly synchronized expression with the rest of the genes, and are expressed in most tissues, being overrepresented among housekeeping genes. Thus, essential genes are topologically and functionally central.
This unexpected peripherality of most disease genes can be best explained by using an evolutionary argument. Mutations in topologically central, widely expressed genes are more likely to result in severe impairment of normal developmental and/or physiological function, leading to lethality in utero or early extrauterine life and to eventual deletion from the population. Only mutations compatible with survival into the reproductive years are likely to be maintained in a population. Therefore, disease-related mutations in the functionally and topologically peripheral regions of the cell give a higher chance of viability.
Disease genes whose mutations are somatic should not be subject to the selective pressure discussed above. Instead, somatic mutations that lead to severe disease phenotypes should more likely affect the functional center. To test the predictive power of this selection-based argument, we studied separately the properties of somatic cancer genes (Cancer Genome Census;
www.sanger.ac.uk/genetics/CGP/Census) and found that they (
i) are more likely to encode hubs, (
ii) show higher coexpression with the rest of the genes in the cell, and (
iii) are more represented among housekeeping genes (
SI Fig. 10). The observed functional and topological centrality of somatic cancer genes fits well with our current understanding that many cancer genes play critical roles in cellular development and growth (
11).